

【聚合氯化鋁】
眾所周知,磷是導(dǎo)致水體富營養(yǎng)化的重要元素之一.據(jù)報道,磷酸陰離子在水體中的含量超過2 μmol ·L-1時,水體便會出現(xiàn)不同程度的富營養(yǎng)化.同時,磷也是一種生態(tài)系統(tǒng)不可或缺的稀缺資源,而且難以更新.因此,實現(xiàn)污染水體中磷的去除并加以回收或再利用至關(guān)重要.目前,很多新型技術(shù)(如膜過濾法、化學(xué)沉淀法、吸附法等)被應(yīng)用于水體中磷的捕集與回收.在眾多方法中,吸附法一直被公認(rèn)為是去除和回收水體中磷的*有效方法之一.對于吸附技術(shù)而言,優(yōu)異的吸附劑開發(fā)一直是該方法的核心.
近年來,粉煤灰、樹脂、生物質(zhì)炭以及幾種金屬氧化物等被廣泛用于磷的吸附.其中,由廢棄生物質(zhì)經(jīng)高溫、限氧熱解所制備的生物質(zhì)炭,因其制備過程簡單,前驅(qū)體來源廣泛等優(yōu)勢而備受關(guān)注.當(dāng)前,各類生物質(zhì)炭被不斷地應(yīng)用于含磷廢水的處理.如:Jung等考察了700℃時以花生殼為前驅(qū)體制備的生物質(zhì)炭對磷吸附的特性,取得了較好的結(jié)果,且由Langmuir模型計算出的*大理論磷吸附容量為7.57 mg ·g-1;Mor等采用稻殼為原料,在500℃限氧熱解制備出的生物質(zhì)炭,對磷的理論*大吸附容量為0.74 mg ·g-1;Takaya等采用橡木為原料,在600℃下限氧熱解制備出的生物質(zhì)炭,對磷的理論*大吸附容量為3.60 mg ·g-1.可見,生物質(zhì)炭雖能有效去除廢水中的磷,但普遍存在吸附容量低等阻礙生物質(zhì)炭推廣應(yīng)用的技術(shù)障礙.不同于生物質(zhì)炭,氧化鎂的Zeta電位處于12,可與磷酸陰離子形成特定的化合物,從而對磷表現(xiàn)出很高的吸附容量和吸附專屬性.然而,氧化鎂通常以粉末形式存在,顆粒尺寸很小,實際應(yīng)用時的固液分離成為其難以逾越的技術(shù)鴻溝.
本研究以花生殼在高溫限氧條件下制備的生物質(zhì)炭為載體,采用氧化鎂作為活性成分對其進(jìn)行改性,研制出了氧化鎂基生物質(zhì)炭復(fù)合材料(MgO-BC).氧化鎂的引入有效地提升了原生生物質(zhì)炭對磷的吸附容量和吸附選擇性,而生物質(zhì)炭的存在又大大提升了氧化鎂應(yīng)用時的固液分離特性.本文首先根據(jù)磷的吸附能力優(yōu)化了材料的制備過程,然后系統(tǒng)地考察了*優(yōu)條件下制備的MgO-BC對中磷的吸附特性受溶液pH、接觸時間、共存離子等因素的影響,并通過遠(yuǎn)紅外光譜技術(shù)對MgO-BC吸附P(V)的機(jī)制進(jìn)行了探討.
1 材料與方法1.1 試劑與儀器
所有試劑均為分析純,均購于上海晶純生化科技股份有限公司,實驗用水為超純水.
1 g ·L-1的磷儲備液由固體KH2PO4溶解制得.花生殼取自于杭州市的大型農(nóng)貿(mào)市場.主要實驗儀器包括:HZ-9310KB恒溫生化搖床(太倉市華利達(dá)實驗設(shè)備有限公司),pH計,烘箱,TU-1810紫外分光光度計(北京普析通用儀器有限責(zé)任公司),OTF-1200x真空管式高溫?zé)Y(jié)爐(合肥科晶材料技術(shù)有限公司),Supra-55掃描電子顯微鏡顯微鏡(德國),Nicolet 6700 FTIR傅里葉變換紅外光譜儀(美國),STA-409PC同步熱分析儀(德國),ASAP2020型全自動微孔物理化學(xué)吸附儀(美國),X'Pert Powder型X射線衍射儀(荷蘭).
1.2 MgO-BC的制備
MgO-BC以粒徑0.154~1.25 mm的花生殼為母體合成,所用方法為高溫限氧煅燒技術(shù).首先將選取的花生殼反復(fù)用自來水清洗去除表面污垢、灰塵,接著用純水沖洗3次并于333 K條件下烘干至恒重.烘干后的花生殼研磨、過篩(0.154~1.25 mm),選出所需粒徑的花生殼備用.
將1 g干態(tài)花生殼加入到100 mL一定濃度的MgCl2溶液中,并用磁力攪拌器持續(xù)攪拌24 h.然后,蒸發(fā)結(jié)晶,將獲得的固體物質(zhì)置于管式爐中,在Ar氣氛圍下熱解.*后,取出熱解后的產(chǎn)物研磨、過篩(16~100目),選取粒徑0.154~1.25 mm的顆粒備用.
1.3 靜態(tài)吸附實驗
準(zhǔn)確稱取一定量的MgO-BC于100 mL具塞玻璃錐形瓶中,分別加入50 mL一定濃度的P溶液,并采用1.0 mol ·L-1、0.1 mol ·L-1的HCl或NaOH溶液控制體系pH值.然后,將錐形瓶置于恒溫振蕩器內(nèi)振蕩12 h,轉(zhuǎn)速180 r ·min-1,反應(yīng)溫度298 K.競爭實驗中,在溶液內(nèi)加入一定量的NaCl、NaNO3、NaHCO3、Na2SO4作為背景離子,考察MgO-BC選擇性吸附P的能力;而對于動力學(xué)實驗,每隔一定時間從250 mL的P溶液內(nèi)取出1.0 mL溶液用于測定P的去除率隨時間的變化規(guī)律.振蕩結(jié)束后,濾除MgO-BC,測定溶液中P的平衡濃度(mg·L-1),平衡吸附量通過式(1) 計算:
(1)
式中,qe表示平衡吸附量(mg ·g-1),V表示P溶液的體積(L),m為吸附材料的質(zhì)量(g),c0為P的初始濃度(mg ·L-1).
1.4 分析方法
溶液中P的濃度使用分光光度計進(jìn)行測定,測定方法為鉬酸銨分光光度法.室溫下放置15 min后,使用光程為30 mm比色皿,在700 nm波長下,以水做參比,測定吸光度.每個樣品吸光度重復(fù)測試3次,*終取平均值.
1.5 表征方法
MgO-BC的表面形貌結(jié)構(gòu)采用掃描電子顯微鏡(Supra-55,Carl Zeiss,德國)進(jìn)行觀察;MgO-BC的熱穩(wěn)定性使用同步熱分析儀(STA409PC,德國)測定,升溫速率為10℃ ·min-1, 采用N2為載氣;MgO顆粒的晶型采用X射線衍射儀(X'Pert Powder,荷蘭)進(jìn)行分析,掃描速度為0.02(°) ·s-1,晶粒尺寸的計算使用Jade 6.0軟件;MgO-BC表面的官能團(tuán)信息采用傅里葉紅外變換光譜儀(Nicolet 6700,美國)分析獲得,測量的波數(shù)范圍在400~4 000 cm-1;MgO-BC的氮氣吸附-解吸實驗借助于N2吸附儀(Micromeritics ASAP2020,美國),在77K條件下測試,利用BET模型獲得比表面積,利用BJH模型獲得孔徑分布結(jié)果.
2 結(jié)果與討論2.1 合成條件的優(yōu)化
本研究從MgCl2的濃度、煅燒溫度和煅燒時間幾方面探究材料的*優(yōu)制備條件(以磷的吸附能力為參考標(biāo)準(zhǔn)).復(fù)合材料對P(V)的吸附效果隨制備參數(shù)的變化情況如圖 1所示.由圖 1(a)可得,300~700℃煅燒溫度下制備的MgO-BC對磷的吸附容量并無顯著差別,但考慮MgCl2只有在溫度高于450℃時,才會徹底氧化分解生成MgO晶體,故而本研究選擇600℃作為*佳燃燒溫度;根據(jù)圖 1(b),當(dāng)MgCl2的初始濃度高于2.50 g ·L-1時(1 g生物質(zhì)炭加入到100 mL MgCl2溶液中),制備的MgO-BC對磷的吸附量達(dá)到*大,本著節(jié)約成本的理念,本文選擇2.5 g ·L-1作為MgCl2的*佳合成濃度;由圖 1(c)可知,在本文研究的煅燒時間(0.5~4.0 h)范圍內(nèi),煅燒時間為2.0 h時制備的MgO-BC吸附P的效果*好.
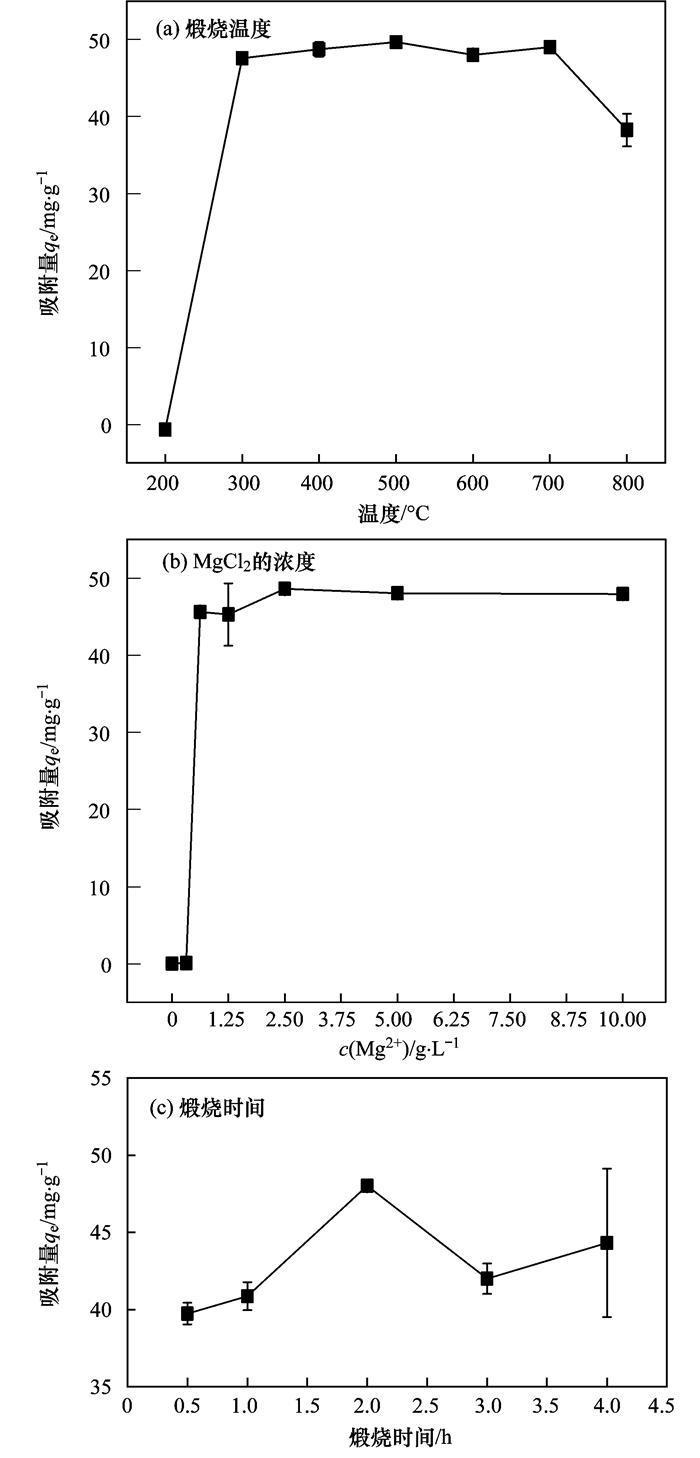
圖 1 煅燒溫度、MgCl2的濃度以及煅燒時間對MgO-BC吸附P的影響
MgO隨溫度的分解過程如下:
(2)
(3)
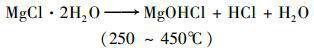
(4)
(5)
綜上所述,若以磷的吸附容量為參考標(biāo)準(zhǔn),則MgO-BC的*佳制備條件為燃燒溫度600℃,MgCl2的濃度2.5 g ·L-1,煅燒時間2.0 h.以下吸附性能評價所用MgO-BC均為該*優(yōu)條件下制備所得.
2.2 BC、MgO和復(fù)合材料MgO-BC的表征
材料的表征是了解材料的形貌特征及物理化學(xué)特性常用的手段.本文對制備的MgO-BC進(jìn)行了一系列表征,為了對比,文中也提供了BC和MgO單體的表征數(shù)據(jù),結(jié)果如圖 2所示.由SEM圖像[圖 2(a)]可知,MgO已被成功負(fù)載于生物質(zhì)炭表面,且載入的MgO顆粒呈針狀晶體結(jié)構(gòu),并均勻地負(fù)載在生物質(zhì)炭表面;而載體生物質(zhì)炭的表面十分粗糙,并含有大量孔狀結(jié)構(gòu),這便為MgO的沉積和負(fù)載提供了有利的條件.從熱重曲線圖[圖 2(b)]可得,當(dāng)溫度升至800℃時,BC的質(zhì)量損失為11%,MgO的質(zhì)量損失僅為4%,復(fù)合材料的質(zhì)量損失僅為8%,這說明經(jīng)高溫煅燒后的復(fù)合材料熱穩(wěn)定性很好,且熱穩(wěn)定性介于BC和MgO之間. MgO-BC吸附P前后的衍射圖譜[圖 2(c)]顯示,復(fù)合材料在衍射角2θ為36.9°(111)、42.9°(200)、62.2°(220)、74.7°(311) 和78.6°(222) 處分別出現(xiàn)不同強(qiáng)度的峰,峰位剛好與MgO的標(biāo)準(zhǔn)圖譜(JCPDSNO.45-0946) 和MgO的衍射圖譜一一對應(yīng),再次說明MgO被成功負(fù)載.尖銳的峰型說明負(fù)載的氧化鎂晶型較好,與SEM圖中MgO的形貌相符,通過德拜-謝樂公式計算出的MgO晶粒尺寸處于27~33 nm之間.從衍射圖譜的形狀上可以看出,復(fù)合材料的衍射圖譜由BC和MgO的衍射圖譜組合而成.由N2吸附-脫附曲線[圖 2(d)]可知,復(fù)合材料相比于N2的吸附過程,N2脫附過程出現(xiàn)回滯現(xiàn)象,存在明顯的H1滯后環(huán),根據(jù)國際純粹化學(xué)組織(IUPAC)的分類,該曲線屬于Ⅳ型等溫線,預(yù)示著復(fù)合材料以介孔為主,這與孔分布的結(jié)果[圖 2(e)]比較相符. BC、MgO的BET比表面積分別為339.2 m2 ·g-1和1.9 m2 ·g-1;復(fù)合材料的BET比表面積為182.3 m2 ·g-1,遠(yuǎn)高于其他的生物質(zhì)炭基復(fù)合材料.
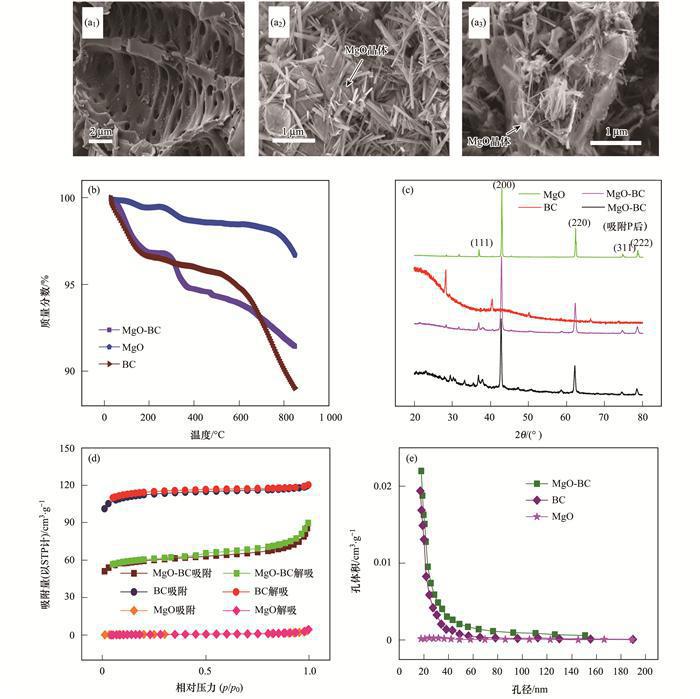
(a1) BC、(a2) MgO和(a3) MgO-BC的SEM圖;(b)BC、MgO和MgO-BC的熱重曲線;(c)BC、MgO和MgO-BC吸附P前后的XRD圖譜;(d)BC、MgO和MgO-BC的N2吸附/脫附等溫線;(e) BC、MgO和MgO-BC的孔徑分布
圖 2 BC、MgO和MgO-BC的各類表征
2.3 pH值的影響
溶液pH通常能夠影響污染物質(zhì)的存在形態(tài)及吸附劑的表面物理化學(xué)性質(zhì).當(dāng)溫度為298 K,P(V)的初始濃度為200 mg ·L-1,固液比為1 g ·L-1時,MgO-BC對P的去除率隨溶液pH值的變化情況如圖 3(a)所示.可見,當(dāng)溶液初始pH值在酸性范圍時(2~7),隨著溶液pH值的升高,P的吸附量不斷增大.其中,溶液pH處于較酸性范圍內(nèi)(2~6) 時,P的去除率隨pH值的升高幾乎呈現(xiàn)直線增長;而溶液初始pH處于弱堿性范圍內(nèi)(7~9) 時,P的去除率隨著pH的升高顯著增加;當(dāng)初始pH處于7~12時,P的去除率隨著pH值的升高而有所下降. MgO的等電點很高,處于pH=12左右[9],因此,當(dāng)溶液pH低于12以下,MgO表面基團(tuán)主要帶正電,而且隨著pH的升高,表面的電負(fù)性變大,這便導(dǎo)致堿性范圍內(nèi)(pH 10~12) 的磷去除率隨著pH的升高而降低.而酸性范圍內(nèi),磷的吸附量隨著pH增加而升高,主要是因為隨著pH的變化磷的形態(tài)轉(zhuǎn)變所致,如圖 3(b),pH處于2~6時,隨著溶液pH值的提升,磷的存在形態(tài)由中性的H3PO4不斷向負(fù)電型的HPO42-/H2PO4-轉(zhuǎn)化,這便導(dǎo)致P吸附量不斷攀升.同時,在較低pH范圍,部分MgO會被酸溶解,進(jìn)入溶液中形成Mg2+,導(dǎo)致了吸附劑表面吸附活性位點的減少,這也導(dǎo)致了酸性范圍內(nèi)磷的吸附隨pH降低而變?nèi)?
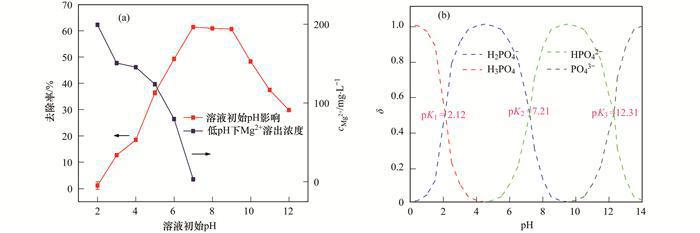
(a)溶液初始pH值對MgO-BC吸附P的影響和低pH條件下Mg溶出濃度;(b)溶液pH值對P存在形態(tài)的影響
圖 3 溶液初始pH值對MgO-BC吸附P的影響
2.4 吸附動力學(xué)
吸附速率是評價吸附劑實際應(yīng)用潛能的一個重要標(biāo)準(zhǔn).快速的吸附過程不僅可減少吸附劑的填裝量,而且可減少固體投資.為了評價復(fù)合材料MgO-BC對P的吸附速率,本文考察了MgO-BC對P的吸附容量隨時間的變化趨勢.如圖 4所示,前100 min吸附速度較快,幾乎呈現(xiàn)直線增長,但隨著時間的推移,吸附速率逐漸降低,并在540 min內(nèi)逐漸達(dá)到吸附平衡.為了進(jìn)一步理解吸附過程,文中還使用了偽一級、偽二級動力學(xué)模型以及粒內(nèi)擴(kuò)散模型對該吸附動力學(xué)曲線進(jìn)行了擬合,見公式(6)~(8).
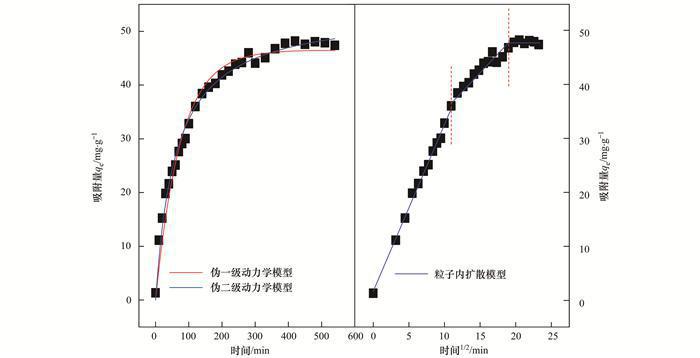
圖 4 MgO-BC吸附P的動力學(xué)曲線
![]()
(6)
(7)
(8)
式中,qe和qt分別表示平衡時和t時刻的吸附量(mg ·g-1),k1表示偽一級動力學(xué)吸附速率常數(shù),k2表示偽二級動力學(xué)吸附速率常數(shù),k3表示粒子內(nèi)擴(kuò)散吸附速率常數(shù).各個模型的擬合參數(shù)結(jié)果見表 1.可見,偽一級和偽二級動力學(xué)模型均能較好地模擬MgO-BC吸附P的動力學(xué)曲線,并且擬合系數(shù)均高于0.97.另外,由偽一級動力學(xué)模型計算出的qe(46.51 mg ·g-1)更接近于實驗值qe(48.21 mg ·g-1).

表 1 MgO-BC吸附P的各種動力學(xué)模型擬合參數(shù)
從顆粒內(nèi)擴(kuò)散模型的擬合結(jié)果可知,MgO-BC吸附P的過程可分為3個階段,這與經(jīng)典的吸附三階段理論吻合.**階段為P從溶液擴(kuò)散至MgO-BC表面,主要受P的濃度梯度控制;第二階段是P由吸附劑表面進(jìn)一步遷移至吸附位點處,主要受吸附劑表面化學(xué)性質(zhì)和孔道結(jié)構(gòu)控制;第三階段即為P發(fā)生吸附的過程,可認(rèn)為瞬間完成.顯而易見,第二階段,即P在復(fù)合物粒內(nèi)擴(kuò)散的過程是該體系的決速步驟,此過程花費時間*多,這是因為MgO-BC的孔道以介孔為主,不利于磷在粒內(nèi)的傳質(zhì)過程.
2.5 競爭吸附實驗
天然水體或工業(yè)廢水中普遍存在著大量環(huán)境友好型的陰離子(如Cl-、NO3-、HCO3-、SO42-等),這些共存離子的濃度通常高出目標(biāo)污染物P數(shù)倍.因此,給定吸附劑能否有效排除此類共存物質(zhì)的干擾直接決定了它們的實際應(yīng)用潛力.為了評價MgO-BC的實際應(yīng)用前景,本研究選擇Cl-、NO3-、HCO3-和SO42-等水體常見陰離子作為共存離子考察了MgO-BC對P的吸附選擇能力,相關(guān)結(jié)果見圖 5.可見,共存離子的引入雖對MgO-BC吸附P產(chǎn)生了一定的不良影響,但當(dāng)競爭離子高于目標(biāo)污染物的10倍時,MgO-BC依然對P保持較高的去除率.另外,4種離子對P吸附的影響能力依次為SO42- NO3- HCO3- Cl-,這與4種離子的交換勢大小順序一致.復(fù)合材料MgO-BC對P的高吸附選擇性,主要是因為溶液中不同形態(tài)的P均可與負(fù)載的氧化鎂形成專屬的作用力,即:
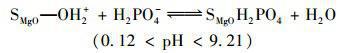
(9)
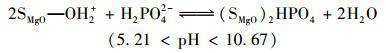
(10)
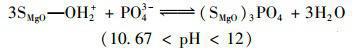
(11)
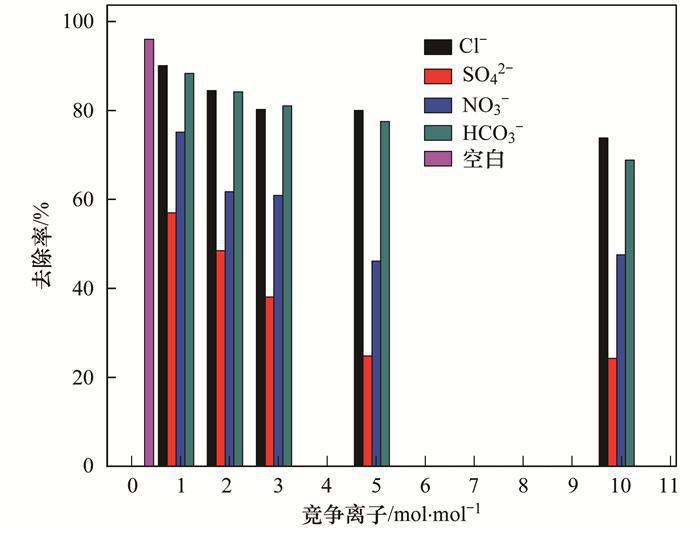
圖 5 不同競爭離子對MgO-BC吸附P的影響
負(fù)載的MgO與P之間形成的特定單核、雙核或者三核的復(fù)合物很好地排除了其他競爭離子的干擾.
2.6 等溫吸附實驗
本研究考察了常溫下(298K)下MgO-BC吸附P的等溫線,結(jié)果如圖 6所示.從中可見,MgO-BC對磷的吸附容量隨著平衡濃度的增加而增大.本文還將該溫度下的等溫線采用經(jīng)典的Langmuir和Freundlich等溫模型進(jìn)行了擬合,擬合參數(shù)見表 2.
(12)
(13)
式中,ce表示P的平衡濃度(mg ·L-1);qe表示P的平衡吸附量(mg ·g-1);qm表示*大吸附容量(mg ·g-1);KL(L ·mg-1),Kf(mg1-n ·Ln ·g-1)以及n均為常數(shù).研究發(fā)現(xiàn),Langmuir模型比Freundlich模型更適合于描述MgO-BC吸附P的過程,且擬合系數(shù)高達(dá)99%,說明P在MgO-BC表面的吸附為單分子層吸附.由Langmuir模型計算出的*大吸附容量為138.07 mg ·g-1,高于實驗值124.83 mg ·g-1.研究還將MgO-BC對P的吸附容量與未改性或改性的生物質(zhì)炭和其他幾種典型吸附劑進(jìn)行了對比,具體結(jié)果見表 3.可見,MgO-BC對磷的吸附量明顯高于其他吸附劑.

圖 6 298K下MgO-BC吸附P的等溫線

表 2 MgO-BC吸附P的等溫線參數(shù)
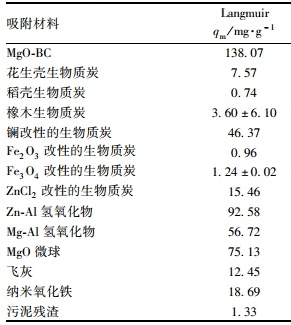
表 3 MgO-BC與其他幾種吸附劑對P的理論*大吸附容量對比
2.7 紅外光譜圖分析
為更深入地揭示MgO-BC吸附水體中P的機(jī)制,本研究還將吸附P前后的MgO-BC進(jìn)行了紅外分析,得到的紅外光譜圖如圖 7所示.從中可知,兩種物質(zhì)的紅外圖譜在3 430 cm-1、1 630 cm-1處均出現(xiàn)了很強(qiáng)的吸收峰,這是樣品中吸收的水分子中羥基振動所致.比較吸附P前后的MgO-BC紅外圖譜發(fā)現(xiàn),吸附P后出現(xiàn)了1 423 cm-1和561 cm-1兩處新的吸收峰,代表P—O鍵,可證實溶液中磷被吸附至復(fù)合材料表面.此外,吸附前的MgO-BC紅外圖譜顯示在波數(shù)1 095 cm-1處出現(xiàn)了明顯的Mg—O鍵吸收峰,從光譜學(xué)角度再次證明氧化鎂被成功負(fù)載于生物質(zhì)炭上,而該吸收峰在磷吸附后遷移至1 071 cm-1處,此處藍(lán)移說明Mg—O鍵參與了磷的吸附過程,且吸附后該鍵作用變強(qiáng).與已有研究對比發(fā)現(xiàn),1 071 cm-1處對應(yīng)的化學(xué)鍵為Mg—O—P[15]鍵,指示著負(fù)載的MgO是復(fù)合材料吸附磷活性位點之一,這與競爭吸附實驗結(jié)果是一致的.
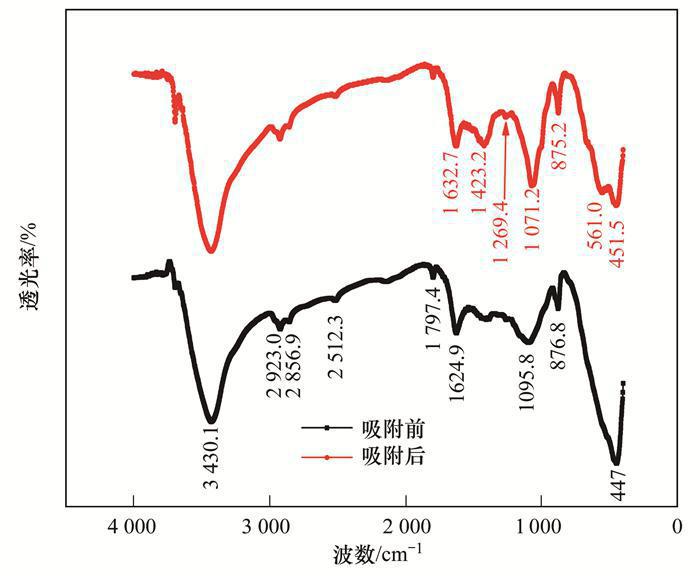
圖 7 吸附P前后的MgO-BC的紅外光譜分析
3 環(huán)境意義
飽和吸附劑的處置是吸附劑應(yīng)用全過程評價的一個重要環(huán)節(jié).本研究擬將吸附飽和后的MgO-BC作為肥料施入土壤.生物質(zhì)炭可以調(diào)節(jié)土壤酸堿度,是很好的土壤改良劑,而Mg是環(huán)境友好元素,不僅對土壤生態(tài)系統(tǒng)無害,且是土壤生物及植物生長必不可少的微量元素之一,MgO-BC中被吸附的磷是一種重要的營養(yǎng)元素,可促進(jìn)土壤作物的生長,且P是隨著Mg復(fù)合物的分解而被不斷釋放,釋放速率較低,不會通過地表徑流污染水體.本研究所設(shè)計的吸附體系不僅可有效降低水體中磷的危害,且實現(xiàn)這種不可再生資源的“變廢為寶”過程,如果實施,還可解決大量農(nóng)業(yè)廢棄物污染環(huán)境的問題,一舉三得.總體而言,該吸附體系符合清潔生產(chǎn)的理念,符合國家提倡的環(huán)保理念,可為其他基于綠色理念設(shè)計的污染控制措施提供一定的借鑒.具體參見資料或更多相關(guān)技術(shù)文檔。
4 結(jié)論
(1) 采用MgO改性BC制備的復(fù)合材料MgO-BC對P具有較好地吸附效果,在pH 7~9范圍內(nèi),吸附效果*好.
(2) MgO-BC吸附P在540 min內(nèi)可達(dá)到吸附平衡,且偽一級和偽二級動力學(xué)方程均能較好地擬合MgO-BC吸附P的動力學(xué)過程,相關(guān)系數(shù)可達(dá)0.973和0.990.
(3) MgO-BC在大量環(huán)境友好型離子共存的條件下,仍具有很強(qiáng)的P吸附選擇性.
(4) MgO-BC吸附P的過程較好地符合Langmuir等溫模型,*大理論吸附容量138.07 mg ·g-1,遠(yuǎn)高于未經(jīng)改性或改性的生物質(zhì)炭和其他幾種典型的P吸附劑.
(5) 吸附飽和后的吸附劑可作為肥料施入土壤,實現(xiàn)磷的循環(huán)利用.

掃一掃,關(guān)注我們












